La enfermedad de Huntington es un trastorno genético fatal que causa la descomposición progresiva de las células nerviosas en el cerebro, tiene un amplio impacto en las capacidades funcionales de una persona y generalmente resulta en trastornos de movimiento, de pensamiento (cognitivos) y psiquiátricos.
La mayoría de las personas desarrollan signos y síntomas en sus 30 o 40 años, pero la enfermedad puede aparecer antes o después en la vida. Cuando la enfermedad se desarrolla antes de los 20 años, la afección se denomina enfermedad de Huntington juvenil, una aparición más temprana de la enfermedad a menudo da como resultado un conjunto de síntomas algo distinta y con una progresión mucho más rápida de la enfermedad.
Hay medicamentos disponibles para ayudar a controlar los síntomas, pero los tratamientos no pueden prevenir el deterioro físico, mental y de comportamiento asociado con la enfermedad. Se conoce como la enfermedad familiar por excelencia porque cada hijo de un padre con esta enfermedad tiene una probabilidad del 50/50 de portar el gen defectuoso.
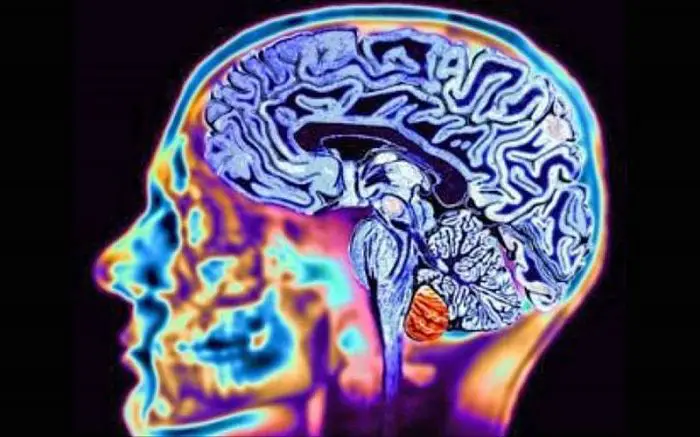
Historia de la enfermedad de Huntington
En 1872, George Huntington describió el desorden en su primer artículo a la edad de 22 años, aunque ha sido reconocido como un trastorno desde al menos la Edad Media, la causa ha sido desconocida hasta hace relativamente poco tiempo. A Huntington se le dieron diferentes nombres a lo largo de esta historia a medida que la comprensión de la enfermedad cambió. Originalmente llamado simplemente ‘corea’ para los movimientos de baile desiguales asociados con la enfermedad, también se ha denominado «corea hereditaria» y «corea progresiva crónica».
La primera mención fue debido a una carta hecha por Charles Oscar Waters, publicada en 1842, donde describió «una forma de corea, vulgarmente llamada magro», que incluye descripciones precisas de corea, su progresión, y la fuerte herencia de la enfermedad. En 1846, Charles Gorman observó cómo la mayor prevalencia parecía ocurrir en regiones localizadas, independientemente de Gorman y Waters, ambos estudiantes de Dunglison en Jefferson Medical College en Filadelfia y Johan Christian Lund también produjo una descripción temprana en 1860, señaló específicamente que en Setesdalen, un valle aislado de montaña en Noruega, había una alta prevalencia de demencia asociada con un patrón de trastornos del movimiento brusco que se daban en familias.
La primera descripción exhaustiva de la enfermedad fue realizada por George Huntington en 1872, al examinar la historia médica combinada de varias generaciones de una familia con síntomas similares, se dio cuenta de que sus condiciones deben estar relacionadas; presentó su definición detallada y precisa de la enfermedad como su primer artículo. Huntington describió el patrón exacto de herencia de la enfermedad autosómica dominante, años antes del redescubrimiento por parte de los científicos de la herencia mendeliana.
William Osler estaba interesado en el trastorno, y quedó impresionado con el trabajo de Huntington, afirmando que «En la historia de la medicina, hay pocos casos en los que una enfermedad ha sido descrita con mayor precisión, más gráfica o breve». El interés continuo de Osler, combinado con su influencia en el campo de la medicina, ayudó a difundir rápidamente la conciencia y el conocimiento del trastorno en toda la comunidad médica.
Causas de la enfermedad de Huntington
La enfermedad de Huntington es causada por un gen defectuoso que se presenta en familias.
Genes y cromosomas
Los genes son las instrucciones de todas las partes del cuerpo humano y el cerebro, están hechos de ADN y empaquetados en hebras llamadas cromosomas, tenemos dos copias de todos nuestros genes, por lo que nuestros cromosomas están en pares. Los humanos tenemos 46 cromosomas (23 pares). El gen defectuoso que causa la enfermedad de Huntington se encuentra en el cromosoma número cuatro.
La copia normal del gen produce una proteína llamada huntingtina, pero el gen defectuoso contiene una región anormal, esta área es más grande de lo normal y produce una forma mutante de huntingtina. Las células en partes del cerebro, específicamente los ganglios basales y partes de la corteza, son muy sensibles a los efectos de la huntingtina anormal, esto los hace funcionar mal y finalmente mueren.
El cerebro normalmente envía mensajes a través de los ganglios basales y la corteza para controlar el movimiento y el pensamiento, así como la motivación, si esta parte del cerebro está dañada, causa problemas con el control del movimiento, el comportamiento y el pensamiento.
Todavía no está claro exactamente cómo la huntingtina anormal afecta las células del cerebro y por qué algunas son más sensibles que otras.
Síntomas de la enfermedad de Huntington

Las características clínicas de la enfermedad pueden incluir problemas psiquiátricos y dificultades con el comportamiento, la alimentación, la comunicación y los movimientos anormales. Las personas pueden comenzar a mostrar las características de la enfermedad de Huntington a casi cualquier edad, pero la mayoría desarrollará problemas entre las edades de 35 y 55 años.
La condición por lo general progresa y empeora por alrededor de 10-25 años, hasta que la persona finalmente muere. Los signos y síntomas pueden variar entre individuos y no hay un patrón típico.
Las características iniciales, como los cambios de personalidad, los cambios de humor y el comportamiento inusual, a menudo se pasan por alto al principio y se atribuyen a otra cosa. Algunas personas con la enfermedad de Huntington pueden no reconocer que tienen algún problema.
Cambios de comportamiento
Los cambios en el comportamiento son a menudo las primeras características que aparecen en la enfermedad y pueden ser las más angustiosas. Estos cambios a menudo incluyen:
- Una falta de emociones y no reconocer las necesidades de los demás en la familia.
- Períodos alternos de agresión, excitación, depresión, apatía, comportamiento asocial y enojo
- Dificultad para concentrarse en más de una tarea y manejar situaciones complejas
- Irritabilidad e impulsividad
Una persona con la enfermedad de Huntington puede carecer de impulso, iniciativa y concentración, lo que los hace parecer vagos. Sin embargo, este no es el caso, es solo la forma en que la condición afecta el cerebro. Como parte de esto, también pueden desarrollar una falta de interés en la higiene y el cuidado personal.
Problemas psiquiátricos
Muchas personas con la enfermedad de Huntington tienen depresión, Esto ocurre como parte de la condición, no solo como una respuesta al diagnóstico. Los síntomas de depresión incluyen un bajo estado de ánimo continuo, baja autoestima, falta de motivación o interés en las cosas y sentimientos de desesperanza.
Algunas personas también pueden desarrollar comportamientos obsesivos y problemas de tipo esquizofrénico, aunque esto es relativamente raro.
Los estudios han demostrado que las personas con la enfermedad de Huntington son más propensas a considerar el suicidio, particularmente cerca del momento del diagnóstico cuando la afección se hace evidente, y cuando comienzan a perder su independencia.
Problemas de movimiento
La enfermedad de Huntington afecta el movimiento. Las características tempranas incluyen movimientos lentos e incontrolables de la cara y sacudidas, o movimientos inquietos de las extremidades y el cuerpo, Estos se mueven de un área del cuerpo a otra y pueden hacer que la persona se tambalee y se tambalee.
Estas características a menudo se ven por primera vez cuando la persona está caminando o descansando (sentado en una silla o acostado en la cama).
A medida que la condición progresa, los movimientos incontrolables serán más frecuentes y extremos. Sin embargo, con el tiempo esto puede cambiar y en las etapas avanzadas de la condición los movimientos de una persona pueden volverse lentos y sus músculos más rígidos.
Problemas de alimentación
Las personas con esta enfermedad tienden a perder peso, a pesar de tener buen apetito, pueden encontrar que comer es agotador, frustrante y desordenado porque los músculos de la boca y la garganta no funcionan correctamente debido a la pérdida de control del motor. En algunos casos, esto puede provocar asfixia e infecciones recurrentes en el pecho.
La pérdida de coordinación puede provocar derrames o caídas de alimentos. La ingestión es un problema, por lo que ahogarte con comida y bebida, especialmente bebidas delgadas como el agua, puede ser un problema común. Una remisión a un dietista o un terapeuta del habla y el lenguaje puede ser necesaria si hay dificultades para tragar, en algunos casos, se puede insertar un tubo de alimentación.
Problemas de comunicación
La comunicación y la cognición (percepción, conciencia, pensamiento y juicio) se ven afectados por la enfermedad de Huntington. Las personas con esta afección a menudo tienen dificultad para expresar sus pensamientos con palabras y hablar mal, pueden entender lo que se dice, pero es posible que no puedan responder o comunicar lo que entienden. Sin embargo, con el tiempo, una persona con la enfermedad se volverá menos receptiva, más retraída y comunicará poco.
Complicaciones de la enfermedad de Huntington
Después del inicio de la enfermedad, las habilidades funcionales empeoran gradualmente con el tiempo, la tasa de progreso y duración varía, el tiempo desde la aparición hasta la muerte suele ser de 10 a 30 años. La enfermedad juvenil de Huntington suele causar la muerte en los 10 años posteriores a la aparición de los síntomas.
La depresión clínica asociada con la enfermedad puede aumentar el riesgo de suicidio, algunas investigaciones sugieren que el mayor riesgo de suicidio ocurre antes de que se haga un diagnóstico y en las etapas intermedias de la enfermedad cuando una persona ha comenzado a perder la independencia.
Finalmente, una persona requiere ayuda con todas las actividades de la vida diaria y la atención. Al final de la enfermedad, es probable que quede confinado a una cama y no pueda hablar, sin embargo, él o ella generalmente puede entender el lenguaje y tiene una conciencia de familiares y amigos.
Las causas comunes de muerte incluyen:
- Neumonía u otras infecciones.
- Lesiones relacionadas con caídas.
- Complicaciones relacionadas con la incapacidad de tragar.
Tratamiento para la enfermedad Huntington
No hay cura para la enfermedad de Huntington, su progreso no se puede revertir o ralentizar, aunque este es el objetivo de muchos proyectos de investigación. Algunas de las características pueden manejarse con medicamentos y terapias, que pueden ser coordinadas por equipos especializados.
Las terapias, como la terapia del habla y del lenguaje y la terapia ocupacional, pueden ayudar con la comunicación y la vida cotidiana. El ejercicio regular también es muy importante. Las personas que están activas tienden a sentirse mucho mejor física y mentalmente que aquellas que no hacen ejercicio, una persona con la enfermedad de Huntington puede tener poca coordinación, pero caminar de forma independiente, con el uso de ayudas para caminar si es necesario, puede marcar la diferencia.
Medicamentos
Los medicamentos para la enfermedad de Huntington se pueden tomar en forma líquida en muchos casos, si es necesario. La mayoría de estos tienen efectos secundarios, como cansancio extremo, sin embargo, a veces puede ser difícil determinar si estos son síntomas de la afección o un resultado de la medicación.
Antidepresivos para tratar la depresión
Los antidepresivos pueden ayudar a mejorar los cambios de humor y tratar la depresión. Incluyen:
- Antidepresivos SSRI, como fluoxetina, citalopram y paroxetina.
- Antidepresivos tricíclicos, como la amitriptilina.
- Otros tipos de antidepresivos, que incluyen mirtazapina, duloxetina y venlafaxina
Los efectos secundarios son los que incluyen:
- Estreñimiento
- Transpiración
- Temblor
- Dificultad para dormir (insomnio)
Estabilizadores del humor para tratar la irritabilidad o cambios de humor
Los estabilizadores del estado de ánimo, en particular la carbamazepina, se pueden considerar como un tratamiento para la irritabilidad. La olanzapina también puede ayudar, junto con el valproato sódico y la lamotrigina. La dosis de carbamazepina debe aumentarse lentamente y controlarse cualquier efecto secundario, no se puede usar durante el embarazo.
Medicación para suprimir los movimientos involuntarios
Los medicamentos enumerados a continuación suprimen los movimientos involuntarios, o corea, que se observan en la enfermedad de Huntington. En el Reino Unido, generalmente se prefieren los medicamentos antipsicóticos.
- Medicación antipsicótica, como olanzapina, sulpirida, risperidona y quetiapina
- Tetrabenazina: reduce la cantidad de dopamina que llega a algunas de las células nerviosas del cerebro
- Benzodiazepinas, como clonazepam y diazepam
La medicación antipsicótica también puede ayudar a controlar los delirios y los arrebatos violentos, sin embargo, pueden tener efectos secundarios graves, como:
- Rigidez
- Sedación
- Temblor
- Movimientos lentos
Diagnostico de la enfermedad de Huntington
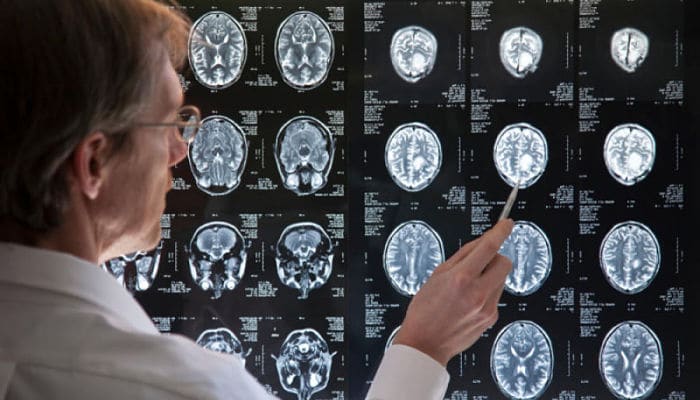
Puede realizarse después de la aparición de los síntomas físicos específicos de la enfermedad. Las pruebas genéticas se pueden usar para confirmar un diagnóstico físico si no hay antecedentes familiares.
Clínico
Sección transversal de un cerebro que muestra tejidos ondulantes con espacios entre ellos, hay dos espacios grandes espaciados equitativamente alrededor del centro.
Sección coronal de un escáner cerebral de RM de un paciente, que muestra atrofia de las cabezas de los núcleos caudados, agrandamiento de los cuernos frontales de los ventrículos laterales y atrofia cortical generalizada
Un examen físico, a veces combinado con un examen psicológico, puede determinar si el inicio de la enfermedad ha comenzado.
Pruebas genéticas predictivas
Debido a que sigue un patrón de herencia autosómico dominante, existe una fuerte motivación para que las personas que están en riesgo de heredarlo busquen un diagnóstico. La prueba genética consiste en una prueba de sangre que cuenta el número de repeticiones en cada uno de los alelos.
Diagnóstico genético preimplantación
Los embriones producidos usando fertilización pueden ser genéticamente evaluados usando diagnóstico genético preimplantacional. Esta técnica, donde se extraen una o dos células de un embrión típicamente de 4 a 8 células y luego se prueba la anomalía genética, se puede utilizar para asegurar que los embriones afectados no se implanten, y por lo tanto cualquier descendencia no heredará la enfermedad.
Pruebas prenatales
También es posible obtener un diagnóstico prenatal para un embrión o feto en el útero, utilizando material genético fetal adquirido a través del muestreo de vellosidades coriónicas, se puede realizar una amniocentesis si el embarazo está más avanzado, dentro de las 14-18 semanas. Este procedimiento analiza el líquido amniótico que rodea al bebé para obtener indicadores de la mutación.
Prevención de la enfermedad de Huntington
Las personas con antecedentes familiares conocidos de la enfermedad de Huntington están comprensiblemente preocupadas sobre si pueden transmitir el gen de Huntington a sus hijos, estas personas pueden considerar pruebas genéticas y opciones de planificación familiar.
Si un padre en riesgo está considerando pruebas genéticas, puede ser útil reunirse con un asesor genético. Un consejero genético discutirá los riesgos potenciales de un resultado positivo de la prueba, lo que indicaría que el padre desarrollará la enfermedad, además, las parejas necesitarán tomar decisiones adicionales sobre si tener hijos o considerar alternativas, como las pruebas prenatales para el gen o la fertilización con esperma del donante.
Otra opción para las parejas es la fertilización y el diagnóstico genético previo a la implantación. En este proceso, los óvulos se eliminan de los ovarios y se fecundan con la esperma del padre en un laboratorio, los embriones se analizan para determinar la presencia del gen de Huntington, y solo los que arrojan resultados negativos para el gen se implantan en el útero de la madre.
► Video recomendado: (Enfermedad de Huntington)
Puedes leer también sobre:

